
Recommendation
 based on reviews by Richard Durbin and 2 anonymous reviewers
based on reviews by Richard Durbin and 2 anonymous reviewers
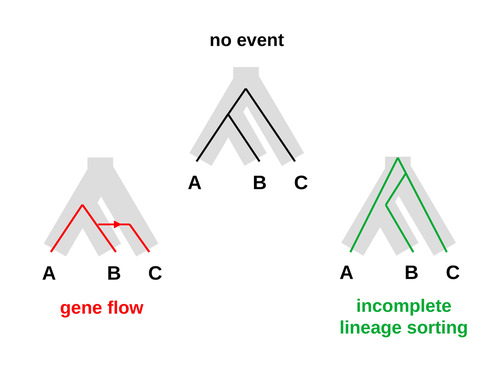
Galtier [1] introduces “Aphid,” a new statistical method that estimates the contributions of gene flow (GF) and incomplete lineage sorting (ILS) to phylogenetic conflict. Aphid is based on the observation that GF tends to make gene genealogies shorter, whereas ILS makes them longer. Rather than fitting the full likelihood, it models the distribution of gene genealogies as a mixture of several canonical gene genealogies in which coalescence times are set equal to their expectations under different models. This simplification makes Aphid far faster than competing methods. In addition, it deals gracefully with bidirectional gene flow—an impossibility under competing models. Because of these advantages, Aphid represents an important addition to the toolkit of evolutionary genetics.
In the interest of speed, Aphid makes several simplifying assumptions. Yet even when these were violated, Aphid did well at estimating parameters from simulated data. It seems to be reasonably robust.
Aphid studies phylogenetic conflict, which occurs when some loci imply one phylogenetic tree and other loci imply another. This happens when the interval between successive speciation events is fairly short. If this interval is too short, however, Aphid’s approximations break down, and its estimates are biased. Galtier suggests caution when the fraction of discordant phylogenetic trees exceeds 50–55%. Thus, Aphids will be most useful when the interval between speciation events is short, but not too short.
Galtier applies the new method to three sets of primate data. In two of these data sets (baboons and African apes), Aphid detects gene flow that would likely be missed by competing methods. These competing methods are primarily sensitive to gene flow that is asymmetric in two senses: (1) greater flow in one direction than the other, and (2) unequal gene flow connecting an outgroup to two sister species. Aphid finds evidence of symmetric gene flow in the ancestry of baboons and also in that of African apes. The data suggest that ancestral humans and chimpanzees both interbred with ancestral gorillas, and at about the same rate. Aphid’s ability to detect this signature sets it apart from competing methods.
References
[1] Nicolas Galtier (2023) “An approximate likelihood method reveals ancient gene flow between human, chimpanzee and gorilla”. bioRxiv, ver. 3 peer-reviewed and recommended by Peer Community in Mathematical and Computational Biology. https://doi.org/10.1101/2023.07.06.547897
DOI or URL of the preprint: https://doi.org/10.1101/2023.07.06.547897
Version of the preprint: 2
, posted 12 Dec 2023, validated 15 Dec 2023See uploaded file.
Download recommender's annotationsDOI or URL of the preprint: https://doi.org/10.1101/2023.07.06.547897
Version of the preprint: 1
, posted 02 Oct 2023, validated 04 Oct 2023See attached
Download recommender's annotationsAs more species have had their genomes sequenced, it has become increasingly clear that there is widespread phylogenetic incongruence between local gene trees and consensus species trees, particularly when the time intervals between successive speciations are relatively small. These incongruences can be caused either by Incomplete Lineage Sorting (ILS) or Gene Flow (GF), and many analytical methods address one or the other, but few both jointly. This paper introduces a new, computationally efficient approach called Aphid to modelling the contribution of both ILS and GF, and with them the times of species separation and basic population genetic parameters of the ancestral species. It appears to me to be novel and effective, giving interesting results, and I recommend publication after revision.
The key step is to greatly simplify the set of possible gene trees considered for explaining the observed data, allowing an efficient maximum likelihood approach to a mixture model over this set. This is very much an intentional heuristic, which results in an enormous reduction in state space, but it appears to be remarkably effective, with good results from reasonable simulations and a demonstration of application to real data. The ideas are nice and the exposition looks sound.
The analysis with Aphid of the human/chimp/gorilla relationships is a potentially important addition to the study of hominine evolution. It suggests that approximately half their genetic discordance is due to gene flow, and consequently that the H-C and HC-G main separation dates are older and ancestral population sizes smaller, which makes sense in a number of ways. There is potential for the author or others to build on this in the future in looking at the relationships of hominine species and subspecies, and considering further the relationship to other data than can be done in this short more technical article.
Like many other phylogenetic approaches, Aphid takes as input a set of supposedly independent gene trees, each built under an assumption of no internal recombination. This referee has general concerns about the no-recombination assumption. For most mammals the average recombination rate is comparable to the mutation rate (e.g. 1e-8 compared to 1.25e-8 for humans) which means that there are on average as many recombination events as mutation events in the ancestral genealogy over a stretch of genome. Given that there have to be mutations present to enable the tree to be defined, then there should also be recombinations. There are many species with smaller mutation rates and much higher recombination rates (because they have smaller chromosomes), such as most invertebrates and many plants, for which the ratio of recombination events to mutations is much higher than one, often an order of magnitude higher. It is true that recombinations are clustered at hotspots, and that neighbouring trees separated by recombinations are correlated, but I would appreciate if you could explicitly discuss the issues for Aphid around the (almost certainly wrong) assumption of no recombination in gene trees.
Major points:
1. L95-101: It took me a while to realise that it was intentional that you are only considering trees with branch lengths at the expected values, rather than all possible trees. There is a good paragraph about this at the end in the discussion, but this modelling decision should be made much clearer earlier on because it is central to your method. First you should say early on that this is a heuristic approach involving major simplification – nothing wrong with that. Then, in the section from lines 95 and following, something more like “We model this set of observed gene trees as coming from a limited mixture of characteristic trees which we call scenarios, which have fixed branch lengths set to the expected values of the branch lengths for that scenario. These fall into three categories.” Then instead of “the coalescence times are assumed to equal” something more like “we model the coalescence times to be fixed at…”. This isn’t really an assumption, because it is flagrantly false – it is a modelling decision.
2. L176: 95% confidence intervals. Are these calculated by re-optimising the likelihood over all the parameters other than the one being investigated for each test value of the parameter under consideration? If so then say this. If not, then this is necessary. Otherwise, if parameters are coupled, it may be that the true confidence intervals are much wider.
a. Related to this, please say for your simulations for what fraction of simulations the true value of each parameter is within the confidence interval, and discuss as appropriate.
b. And you don’t show the confidence intervals in SuppTable2, nor the significance test results. Please add them. Sorry that this adds lots more columns. You could perhaps transpose the table and have columns per data set and rows per feature – your choice.
3. L188-191: how is the root defined for deciding whether trees are imbalanced? Do you need a super-outgroup to set the root, beyond the ones whose lengths from tip to root are being compared to those from? If so, say so. Else explain how this is done. (If you assume ultrametric behaviour to define the root then of course you underestimate root-to-tip variation.)
4. Related to the comments above about the assumption of no recombination within the loci, I would like to see for the real data (Supp Table 2) a new column giving the fraction of 2:2 SNPs involving A,B,C and an outgroup O that is incongruent in the test regions. This is what CoalHMM uses, and is independent of the no-recombination assumption. My memory is that for Human/Chimp/Gorilla/Orang this fraction is 30%, not 26% as you have. If you see such a difference it might help motivate discussion of the consequences of the no-recombination assumption. If you see no difference, then the explanation may be due to lower Ne in exons and/or my faulty memory. In any case, if there is no difference that is a nice validation that the model is behaving reasonably.
5. Your discussion of the real data focuses almost exclusively on macaques and hominins. I think you should at least provide a bit more overview of the other results, otherwise why do them? It looks to me that for horses and mice there is negligible evidence for either GF or ILS – please give the significance test results in the table. For the others, the GF is similar to or greater than ILS. Quite surprising to me and worth remarking on. Is there other literature on these cases?
6. For macaques Song et al. had two M. fascicularis and only one of those shows the strong gene flow signature (the one from Mauritius, where the Portugese introduced M. fascicularis several hundred years ago from SE Asia). They interpret that as meaning that the gene flow has occurred within the last 330k years, since that is their estimated divergence time. However you estimate 63% p_a which is much more ancient. I wonder about the accuracy of your p_a estimate – all the other values are above 90%. You don’t discuss this in your section on simulation. Could you please address how accurately p_a is estimated on the simulation data, and comment on this discrepancy in the macaque analysis.
7. I note that Song et al. inferred bi-directional gene flow in this case, which is possible in principle with careful application of D-stats or 5-taxon tests. I realise that because your method only models symmetric bi-directional gene flow it does not selectively demonstrate bi-directional versus uni-directional gene flow. You should state this somewhere.
8. L343-345: you discuss not testing p_AB. Why not? This would be simple using the same scheme as for p_AC, p_BC. Maybe you have low power for this, but that would be good to report. It would not invalidate the paper at all from my perspective, just show the limits of the approach.
9. L357: why not distinguish N_e(AB) from N_e(ABC)? I would be interested in what happens if you add that to the model. But I realise that this is substantially more work, so I do not require this. If you tried it and it didn’t work well because of indeterminism, I would appreciate a statement to that effect as again being useful to understand the limits of the model, without requiring that you present the results to demonstrate this. In my view it is much more useful to describe things that didn’t work than to bury them. I hope the editor and the other referee(s) take the same view!
Minor points:
1. L24: “These problems are presumably minimized” – this is imprecise. They are presumably less of an issue, but not as small as possible, which is the meaning of minimized. Something more like “these potential problems are presumably much reduced” or “much less of a concern”
2. L76: “J & MR” ref should be Smith and Kronfest.
3. L79: “The ILS hypothesis predicts an exponential distribution for this variable regardless of tree topology” is incorrect. This needs to be “The ILS hypothesis predicts an exponential distribution for this variable for trees discordant with the species tree”, which is what the Edelman paper says.
4. L118: “as from” in place of “than from” is better English
5. L122: You need to state here that you assume at most one GF event.
6. L156: you talk about star topologies here, but later (L192) you rule them out in an indirect way, by saying that you ban trees with internal branches under 0.5 mutations – since the number of mutations is discrete this means with 0 mutations, i.e. star trees. I suggest just to say at L156 that you exclude star topologies with d = 0 (and again at L192).
7. L265: “lead” -> “led”
8. L269: capitalise “Indonesia”
9. Supp Table 2: why is the asymmetry_ILS < 0.5. By definition it should be greater than 0.5, as you say in L174. Also the table header is asymmetry_ILS while the text is imbalance_ILS.
10. L282,L292: you must change “neutral mutation rate” to “exon mutation rate” or even more correctly “exon accepted mutation rate”. By using exons you are clearly not considering neutral sequence.
11. L308: “appears” not “appear” in “appears to exist”
12. L317,L320: “departing from”
13. Figure 3: I find using the magnitude of the disks for the fraction explained hard to evaluate. Fine to leave the disks, but could you add next to them the actually number in text as a percentage (e.g. 12%, 4% etc. – no need for more precision here).
14. L350: “conditional” not “conditionally”
15. Supp Text: “do not coalesce” rather than “do not coalesced”
16. Reference list: Maybe this is an editorial rather than an author point, but for this style (name, year) surely the references should be in alphabetical order of first author.
