
LAMBERT Amaury
- Institute of Biology of the ENS (IBENS), Ecole Normale Supérieure (ENS Paris), Paris, France
- Evolutionary Biology, Genetics and population Genetics, Probability and statistics
- recommender
Recommendations: 2
Reviews: 0
Recommendations: 2
Optimal antimicrobial dosing combinations when drug-resistance mutation rates differ
Optimizing antibiotic use: How much should we favor drugs with lower resistance mutation rates?
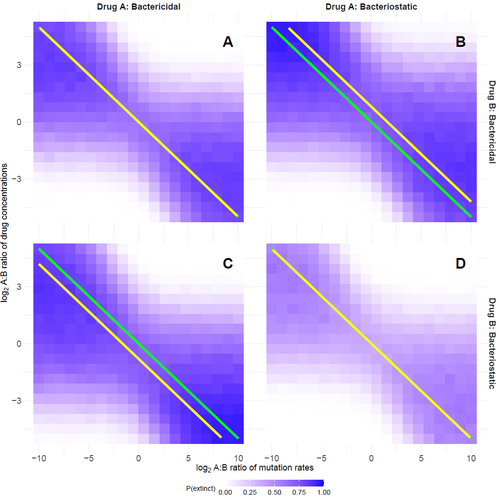
Recommended by Amaury Lambert based on reviews by 2 anonymous reviewersIn the hereby recommended paper [1], Delaney, Letten and Engelstädter study the appearance of antibiotic resistance(s) in a bacterial population subject to a combination of two antibiotics \( A \) and \( B \), in concentrations \( C_A \) and \( C_B \) respectively. Their goal was to find optimal values of \( C_A \) and \( C_B \) that minimize the risk of evolutionary rescue, under the unusual assumption that resistance mutations to either antibiotic are not equally likely.
The authors introduce a stochastic model assuming that the susceptible population grows like a supercritical birth-death process which becomes subcritical in the presence of antibiotics (and exactly critical for a certain concentration c of a single antibiotic): the effect of each antibiotic is either to reduce division rate (bacteriostatic drug) or to enhance death rate (bacteriocidal drug) by a factor which has a sigmoid functional dependence on antibiotic concentration.
Now at each division, a resistance mutation can arise, with probability \( \mu_A \) for a resistance to antibiotic \( A \) and with probability \( \mu_B \) for a resistance to antibiotic \( B \).
The goal of the paper is to find the optimal ratio of drug concentrations \( C_A \) and \( C_B \) when these are subject to a constrain \( C_A + C_B = c \), depending on the ratio of mutation rates. Assuming total resistance and no cross resistance, the authors show that the optimal concentrations are given by \( C_A = c/(1+\sqrt{\mu_A/\mu_B }) \) and \( C_B = c/(1+\sqrt{\mu_B/\mu_A}) \), which leads to the beautiful result that the optimal ratio \( C_A/C_B \) is equal to the square root of \( \mu_B/\mu_A \).
The authors have made a great job completing their initial submission by simulations of model extensions, relaxing assumptions like single antibiotic mode, absence of competition, absence of cost of resistance, sharp cutoff in toxicity… and comparing the results obtained by simulation to their mathematical result.
The paper is very clearly written and any reader interested in antibiotic resistance, stochastic modeling of bacterial populations and/or evolutionary rescue will enjoy reading it. Let me thank the authors for their patience and for their constant willingness to comply with the reviewers’ and recommender’s demands during the reviewing process.
Reference
Oscar Delaney, Andrew D. Letten, Jan Engelstaedter (2025) Optimal antimicrobial dosing combinations when drug-resistance mutation rates differ. bioRxiv, ver.3 peer-reviewed and recommended by PCI Mathematical and Computational Biology https://doi.org/10.1101/2024.05.04.592498
Cancer phylogenetic tree inference at scale from 1000s of single cell genomes
Phylogenetic reconstruction from copy number aberration in large scale, low-depth genome-wide single-cell data.
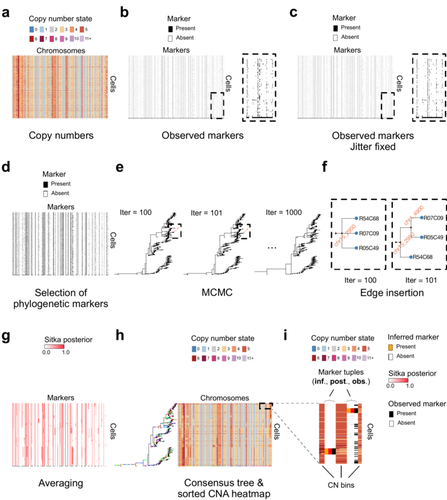
Recommended by Amaury Lambert based on reviews by 3 anonymous reviewersThe paper [1] presents and applies a new Bayesian inference method of phylogenetic reconstruction for multiple sequence alignments in the case of low sequencing coverage but diverse copy number aberrations (CNA), with applications to single cell sequencing of tumors.
The idea is to take advantage of CNA to reconstruct the topology of the phylogenetic tree of sequenced cells in a first step (the `sitka' method), and in a second step to assign single nucleotide variants (SNV) to tree edges (and then calibrate their lengths) (the `sitka-snv' method).
The data are summarized into a binary-valued CxL matrix Y, where C is the number of cells and L is the number of loci (here, loci are segments of prescribed length called `bins'). The entry of Y at row i and column j is 1 (otherwise 0) iff in the ancestral lineage of cell i, at least one genomic rearrangement has occurred, and more specifically the gain or loss of a segment with at least one endpoint in locus j or in locus j+1. The authors expect the infinite-allele assumption to approximately hold (i.e., that at most one mutation occurs at any given marker and that 0 is the ancestral state). They refer to this assumption as the `perfect phylogeny assumption'. By only recording from CNA events the endpoints at which they occur, the authors lose the information on copy number, but they gain the assumption of independence of the mutational processes occurring at different sites, which approximately holds for CNA endpoints.
The goal of sitka is to produce a posterior distribution on phylogenetic trees conditional on the matrix Y , where here a phylogenetic tree is understood as containing the information on 1) the topology of the tree but not its edge lengths, and 2) for each edge, the identity of markers having undergone a mutation, in the sense of the previous paragraph.
The results of the method are tested against synthetic datasets simulated under various assumptions, including conditions violating the perfect phylogeny assumption and compared to results obtained under other baseline methods. The method is extended to assign SNV to edges of the tree inferred by sitka. It is also applied to real datasets of single cell genomes of tumors.
The manuscript is very well-written, with a high degree of detail. The method is novel, scalable, fast and appears to perform favorably compared to other approaches. It has been applied in independent publications, for example to multi-year time-series single-cell whole-genome sequencing of tumors, in order to infer the fitness landscape and its dynamics through time, see [2].
The reviewing process has taken too long, mainly because of other commitments I had during the period and to the difficulty of finding reviewers. Let me apologize to the authors and thank them for their patience as well as for the scientific rigor they brought to their revisions and answers to reviewers, who I also warmly thank for their quality work.
REFERENCES
[1] Sohrab Salehi, Fatemeh Dorri, Kevin Chern, Farhia Kabeer, Nicole Rusk, Tyler Funnell, Marc J Williams, Daniel Lai, Mirela Andronescu, Kieran R. Campbell, Andrew McPherson, Samuel Aparicio, Andrew Roth, Sohrab Shah, and Alexandre Bouchard-Côté. Cancer phylogenetic tree inference at scale from 1000s of single cell genomes (2023). bioRxiv, 2020.05.06.058180, ver. 4 peer-reviewed and recommended by Peer Community in Mathematical and Computational Biology.
https://doi.org/10.1101/2020.05.06.058180
[2] Sohrab Salehi, Farhia Kabeer, Nicholas Ceglia, Mirela Andronescu, Marc J. Williams, Kieran R. Campbell, Tehmina Masud, Beixi Wang, Justina Biele, Jazmine Brimhall, David Gee, Hakwoo Lee, Jerome Ting, Allen W. Zhang, Hoa Tran, Ciara O’Flanagan, Fatemeh Dorri, Nicole Rusk, Teresa Ruiz de Algara, So Ra Lee, Brian Yu Chieh Cheng, Peter Eirew, Takako Kono, Jenifer Pham, Diljot Grewal, Daniel Lai, Richard Moore, Andrew J. Mungall, Marco A. Marra, IMAXT Consortium, Andrew McPherson, Alexandre Bouchard-Côté, Samuel Aparicio & Sohrab P. Shah. Clonal fitness inferred from time-series modelling of single-cell cancer genomes (2021). Nature 595, 585–590. https://doi.org/10.1038/s41586-021-03648-3