
PIBIRI Giulio Ermanno
- Department of Environmental Sciences, Informatics and Statistics, Ca' Foscari University of Venice, Venice, Italy
- Combinatorics, Computational complexity, Design and analysis of algorithms
- recommender
Recommendations: 2
Reviews: 0
Recommendations: 2
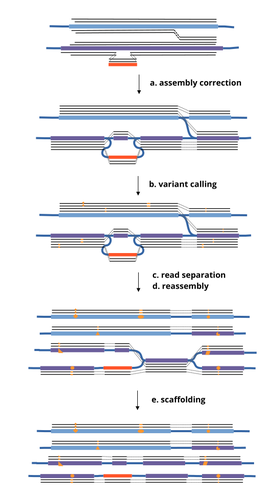
HairSplitter: haplotype assembly from long, noisy reads
Accurate Haplotype Reconstruction from Long, Error-Prone, Reads with HairSplitter
Recommended by Giulio Ermanno Pibiri based on reviews by Dmitry Antipov and 1 anonymous reviewerA prominent challenge in computational biology is to distinguish microbial haplotypes -- closely related organisms with highly similar genomes -- due to small genomic differences that can cause significant phenotypic variations. Current genome assembly tools struggle with distinguishing these haplotypes, especially for long-read sequencing data with high error rates, such as PacBio or Oxford Nanopore Technology (ONT) reads. While existing methods work well for either viral or bacterial haplotypes, they often fail with low-abundance haplotypes and are computationally intensive.
This work by Faure, Lavenier, and Flot [1] introduces a new tool -- HairSplitter -- that offers a solution for both viral and bacterial haplotype separation, even with error-prone long reads. It does this by efficiently calling variants, clustering reads into haplotypes, creating new separated contigs, and resolving the assembly graph. A key advantage of HairSplitter is that it is entirely parameter-free and does not require prior knowledge of the organism's ploidy. HairSplitter is designed to handle both metaviromes and bacterial metagenomes, offering a more versatile and efficient solution than existing tools, like stRainy [2], Strainberry [3], and hifiasm-meta [4].
References
[1] Roland Faure, Dominique Lavenier, Jean-François Flot (2024) HairSplitter: haplotype assembly from long, noisy reads. bioRxiv, ver.3 peer-reviewed and recommended by PCI Math Comp Biol https://doi.org/10.1101/2024.02.13.580067
[2] Kazantseva E, A Donmez, M Pop, and M Kolmogorov (2023). stRainy: assembly-based metagenomic strain phasing using long reads. Bioinformatics. https://doi.org/10.1101/2023.01.31.526521
[3] Vicedomini R, C Quince, AE Darling, and R Chikhi (2021). Strainberry: automated strain separation in low complexity metagenomes using long reads. Nature Communications, 12, 4485. ISSN: 2041-1723. https://doi.org/10.1038/s41467-021-24515-9
[4] Feng X, H Cheng, D Portik, and H Li (2022). Metagenome assembly of high-fidelity long reads with hifiasm-meta. Nature Methods, 19, 1–4. https://doi.org/10.1038/s41592-022-01478-3
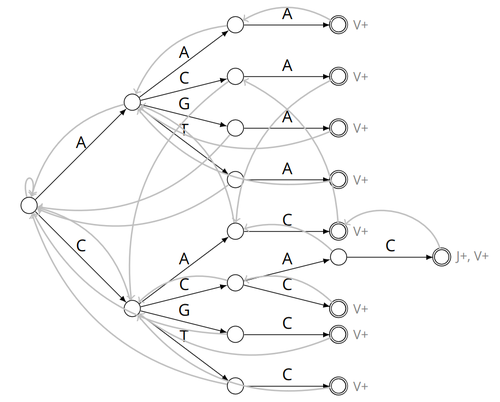
Alignment-free detection and seed-based identification of multi-loci V(D)J recombinations in Vidjil-algo
An accelerated Vidjil algorithm: up to 30X faster identification of V(D)J recombinations via spaced seeds and Aho-Corasick pattern matching
Recommended by Giulio Ermanno Pibiri based on reviews by Sven Rahmann and 1 anonymous reviewerVDJ recombination is a crucial process in the immune system, where a V (variable) gene, a D (diversity) gene, and a J (joining) gene are randomly combined to create unique antigen receptor genes. This process generates a vast diversity of antibodies and T-cell receptors, essential for recognizing and combating a wide array of pathogens. By identifying and quantifying these VDJ recombinations, we can gain a deeper and more precise understanding of the immune response, enhancing our ability to monitor and manage immune-related conditions.
It is therefore important to develop efficient methods to identify and extract VDJ recombinations from large sequences (e.g., several millions/billions of nucleotides). The work by Borée, Giraud, and Salson [2] contributes one such algorithm. As in previous work, the proposed algorithm employs the Aho-Corasick automaton to simultaneously match several patterns against a string but, differently from other methods, it also combines the efficiency of spaced seeds. Working with seeds rather than the original string has the net benefit of speeding up the algorithm and reducing its memory usage, sometimes at the price of a modest loss in accuracy. Experiments conducted on five different datasets demonstrate that these features grant the proposed method excellent practical performance compared to the best previous methods, like Vidjil [3] (up to 5X faster) and MiXCR [1] (up to 30X faster), with no quality loss.
The method can also be considered an excellent example of a more general trend in scalable algorithmic design: adapt "classic" algorithms (in this case, the Aho-Corasick pattern matching algorithm) to work in sketch space (e.g., the spaced seeds used here), trading accuracy for efficiency. Sometimes, this compromise is necessary for the sake of scaling to very large datasets using modest computing power.
References
[1] D. A. Bolotin, S. Poslavsky, I. Mitrophanov, M. Shugay, I. Z. Mamedov, E. V. Putintseva, and D. M. Chudakov (2015). "MiXCR: software for comprehensive adaptive immunity profiling." Nature Methods 12, 380–381. ISSN: 1548-7091. https://doi.org/10.1038/nmeth.3364
[2] C. Borée, M. Giraud, M. Salson (2024) "Alignment-free detection and seed-based identification of multi-loci V(D)J recombinations in Vidjil-algo". https://hal.science/hal-04361907v2, version 2, peer-reviewed and recommended by Peer Community In Mathematical and Computational Biology.
[3] M. Giraud, M. Salson, M. Duez, C. Villenet, S. Quief, A. Caillault, N. Grardel, C. Roumier, C. Preudhomme, and M. Figeac (2014). "Fast multiclonal clusterization of V(D)J recombinations from high-throughput sequencing". BMC Genomics 15, 409. https://doi.org/10.1186/1471-2164-15-409.