
Accurate Haplotype Reconstruction from Long, Error-Prone, Reads with HairSplitter
 based on reviews by Dmitry Antipov and 1 anonymous reviewer
based on reviews by Dmitry Antipov and 1 anonymous reviewer
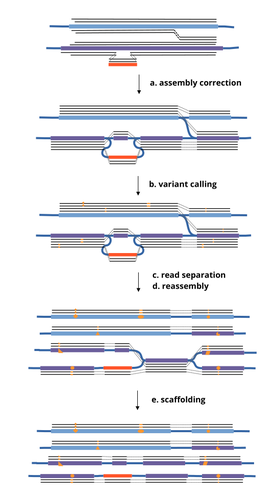
HairSplitter: haplotype assembly from long, noisy reads
Abstract
Recommendation: posted 01 October 2024, validated 02 October 2024
Pibiri, G. (2024) Accurate Haplotype Reconstruction from Long, Error-Prone, Reads with HairSplitter. Peer Community in Mathematical and Computational Biology, 100307. 10.24072/pci.mcb.100307
Recommendation
A prominent challenge in computational biology is to distinguish microbial haplotypes -- closely related organisms with highly similar genomes -- due to small genomic differences that can cause significant phenotypic variations. Current genome assembly tools struggle with distinguishing these haplotypes, especially for long-read sequencing data with high error rates, such as PacBio or Oxford Nanopore Technology (ONT) reads. While existing methods work well for either viral or bacterial haplotypes, they often fail with low-abundance haplotypes and are computationally intensive.
This work by Faure, Lavenier, and Flot [1] introduces a new tool -- HairSplitter -- that offers a solution for both viral and bacterial haplotype separation, even with error-prone long reads. It does this by efficiently calling variants, clustering reads into haplotypes, creating new separated contigs, and resolving the assembly graph. A key advantage of HairSplitter is that it is entirely parameter-free and does not require prior knowledge of the organism's ploidy. HairSplitter is designed to handle both metaviromes and bacterial metagenomes, offering a more versatile and efficient solution than existing tools, like stRainy [2], Strainberry [3], and hifiasm-meta [4].
References
[1] Roland Faure, Dominique Lavenier, Jean-François Flot (2024) HairSplitter: haplotype assembly from long, noisy reads. bioRxiv, ver.3 peer-reviewed and recommended by PCI Math Comp Biol https://doi.org/10.1101/2024.02.13.580067
[2] Kazantseva E, A Donmez, M Pop, and M Kolmogorov (2023). stRainy: assembly-based metagenomic strain phasing using long reads. Bioinformatics. https://doi.org/10.1101/2023.01.31.526521
[3] Vicedomini R, C Quince, AE Darling, and R Chikhi (2021). Strainberry: automated strain separation in low complexity metagenomes using long reads. Nature Communications, 12, 4485. ISSN: 2041-1723. https://doi.org/10.1038/s41467-021-24515-9
[4] Feng X, H Cheng, D Portik, and H Li (2022). Metagenome assembly of high-fidelity long reads with hifiasm-meta. Nature Methods, 19, 1–4. https://doi.org/10.1038/s41592-022-01478-3

The recommender in charge of the evaluation of the article and the reviewers declared that they have no conflict of interest (as defined in the code of conduct of PCI) with the authors or with the content of the article. The authors declared that they comply with the PCI rule of having no financial conflicts of interest in relation to the content of the article.
Ph.D. AMX grant
Evaluation round #2
DOI or URL of the preprint: https://doi.org/10.1101/2024.02.13.580067
Version of the preprint: 2
Author's Reply, 21 Sep 2024
Decision by Giulio Ermanno Pibiri, posted 23 Jul 2024, validated 23 Jul 2024
Dear authors,
both reviewers are satisfied with the modifications apported. Reviewer 1 has no further comments, except for a sentence that you may consider to include in the main body of the manuscript. Reviewer 2, instead, is wondering why you don't comment on the aspect of correctness and contiguity in the Results or Discussion section (in addition to some minor observations). In particular, he is pointing out several misassemblies in one of the tables that can affect downstream analyses.
I think this work merits a minor revision to address the comments of Reviewer 2.
Thank you and I hope to see your revised manuscript soon.
Reviewed by anonymous reviewer 1, 20 Jun 2024
The authors have addressed my comments.
In the response, they provided the sentence "Long indels are treated as multiple adjacent loci." They could also add it to the main text to make it clearer.
Reviewed by Dmitry Antipov, 08 Jul 2024
During the revision authors made great job on improving both text and tool itself.
However there are still some moments where improvement is possible:
Major:
Results still barely mention anything except completeness (I found only one sentence "Particularly with Nanopore data, HairSplitter produced the most complete assemblies, though less contiguous than those produced by Strainbery."). Focus on the completeness metrics is clear, but both correctness and contiguity deserves more attention. I.e. there are hundreds of misassemblies for some datasets in supplementary table 3, and if they are because of the regions of different strains assembled into chimeric contigs this definitely can affect downstream analysis and should be mentioned in the Results or Discussion
Minor:
Possibly this is biorxiv bug but still - there's some mess whether tables 2-5 are suplementary or not. pdf version is consistent, and text refers them as suplementary but web https://www.biorxiv.org/content/10.1101/2024.02.13.580067v2.full shows them in the main text.
Line 118: contigs of the _completed_ assembly? Likely so, but not 100% clear
Line 336: high duplication ratio reference to sup table2 can be beneficial here
Line 294-295: phasing of polyploid organisms
Cited paper was published before the current age of T2T assemblers (hifiasm, verkko) and do not distinguish diploid and polyploid organisms. That assemblers do not have significant problems in separating haplotypes (for polyploids there's still a problem with phasing but in different sence - utilization of Hi-C or other long distance technologies and not in the long read level). So additional motivation for extending hairsplitter to polyploids would be beneficial.
Lines 255, 258: suplementary table 4 instead of 5? Also suggest to add mention about Strainline crash to the caption of that table too.
metaMDBG removal - I get the motivation, but since it is a popular tool would be nice to explain it in the text too
Evaluation round #1
DOI or URL of the preprint: https://doi.org/10.1101/2024.02.13.580067
Version of the preprint: 1
Author's Reply, 17 Jun 2024
Decision by Giulio Ermanno Pibiri, posted 22 Apr 2024, validated 23 Apr 2024
Both reviewers indentify substantial merits of the submitted work. In particular, Reviewer 1 has some minor reservations regarding terminology and missing explanations. Reviewer 2, instead, asks for a better discussion about correctness and completeness of the method.
This preprint merits a revision to adress the comments of the reviewers. Apart for some minor editorial modifications (like captions, terminology, and citation fixes), two major things are required in a revised version of the manuscript:
- Discuss what part is novel and what components are re-used from the state of the art (Reviewer 1).
- Discuss also the aspect of assembly contituity and not only that of completeness (Reviewer 2). Perhaps, some more experiments illustrating the trade-off between contiguity, correctness, and completeness offered by HairSplitter could be presented.
Reviewed by anonymous reviewer 1, 27 Mar 2024
SUMMARY
The authors provide a software HairSplitter to separate (phase) assemblies into strain-haplotypes using a strain-oblivious assembly and ONT or PacBio HiFi reads as input. As highly similar strains have been shown to have very different functional roles, software for accurate strain-specific assembly is needed. Several tools already exist for this (reported by the authors). However, the authors' software substantially improves over other state-of-the-art methods for noisier ONT reads, while performing reasonably well on PacBio HiFi reads.
I find the paper easy to follow and digest (with the exceptions listed in significant comments 1 and 2). The methods, while being succinctly presented, are well described. In particular, the variant calling procedure is simple and elegantly described.
Furthermore, the experiments are well performed against state-of-the-art using both simulated and biological data. The results are clearly presented and easy to follow. HairSplitter's limitations, such as requiring spanning at least five polymorphic loci, are adequately discussed. Two future directions of this work are also described and seem reasonable.
I only have minor-type comments of various significance.
SIGNIFICANT
1. Terminology: The authors use "a new read clustering algorithm" in the abstract, but clustering is not used in the paper. Instead, terms like 'read separation' is used in the figure 1, and 'read binning' is used as a section header. Are they all referring to the same thing? Also, 'scaffolding' (step e in Figure 1) is missing in the text. Is scaffolding the 'duplication' procedure described in the reassembly section? Please address this and use the same terms whenever possible.
2. I was left wondering what methods were novel in this paper and what was re-used. The authors mention that the variant calling procedure is based on an already-explored idea (Z Feng et al., 2021). However, the methods in Z Feng et al., 2021 are quite dense to read. The authors should describe in more detail the similarities/differences to the approach (Z Feng et al., 2021). Also, if the read clustering(/binning) is novel, it could be emphasized not only in the abstract but also in the text.
MINOR
- It should be mentioned what the input and output formats of HairSplitter are at some point early in the paper (e.g., GFA/fasta assembly format and reads in fastq?).
- The authors' statistical derivation for variant calling is elegant, both in the approach and in its simple presentation, allowing the reader to quickly absorb the approach. In addition to significant comment 2, I had some minor comments on it:
- Do the authors differentiate between indels and substitutions? What about indels spanning more than one position in the pileup?
- It would be preferable if the authors denoted the error (currently e) to any other letter (e.g., p?). The inattentive reader could confuse e^{ab} with the exponential function.
- The authors mention that "clusters with more than five positions are deemed robust," but their model would be able to "handle" smaller b if the assumptions were valid. I have worked with similar approaches, and in my experience, it is the assumption that 'errors are independent' that typically does not fit biological data (especially indels in homopolymer regions). It is, in my opinion, totally fine from a modeling perspective to make the simplification of independent errors, but it would help if the authors could describe their reason for this lower threshold in more detail beyond "to avoid the inadvertent selection of artifact-prone positions." Is the authors' experience the same as mine with indels and homopolymers?
- I believe 'MetaQUAST completeness' (Figures) and "Genome Fraction %" (Suppl. Tables) are the same data presented twice under different names. I suggest using consistent naming (or removing duplicated data presentation).
- Supplementary Table 2: Is there a missing comma in Hairsplitter's #mismatches per 100kbp? 31944 seems high. It is potentially a copy-paste error since the NGA50 is also 31944 for that experiment.
- Legend of Figure 3 should include "V.fluvialis" dataset: "27-mer completeness, MetaQUAST completeness and run-time of different software on the V.fluvialis and the three Zymo-GMS datasets."
- I had to download a 5.9Gb file for information on how the tools were run (commands and parameters). It could be added to supplementary data or available in some other form separate from the data.
Reviewed by Dmitry Antipov, 21 Apr 2024
Questions:
Does the title clearly reflect the content of the article?
Yes
Does the abstract present the main findings of the study?
Possibly, Not completely convinced (will explain later)
Are the research questions/hypotheses/predictions clearly presented?
Yes
Does the introduction build on relevant research in the field?
Yes
Are the methods and analyses sufficiently detailed to allow replication by other researchers?
Yes
Are the methods and statistical analyses appropriate and well described?
Yes, with minor questions
Are the results described and interpreted correctly?
Not convinced, will explain later
Have the authors appropriately emphasized the strengths and limitations of their study/theory/methods/argument?
Questionable
Are the conclusions adequately supported by the results (without overstating the implications of the findings)?
Questionable
Major issues:
As for me, in the assembly-related paper the most important part is the results section. And here I see a clear problem on focusing on only completeness, with contiguity issues completely ignored in the main text. Supplementary table 3 shows that for some of the strains NA50 of HairSplitter is dramatically lower(6K vs 180K!) than for competitiors or even the original Flye assembly. I'm not sure whether such fragmented assembly, without haplotype labels, makes lots of sence for downstream analysis.
This is quite possible that tradeoff between contiguity, correctness and completeness favors HairSplitter, but this definitely should be better shown and discussed in the main text. As for now I'm not convinced.
Assembly correction stage name is likely misleading.
If significant amount of reads stops aligning, this may be still not a misassembly if another reads align.
Consider two strains, ABC and AB'C
It's quite possible for the assembler to report it as one large contig ABC and additional contig B'. HairSplitter may split ABC into three separate contigs A, B, C since reads from the second strain stop aligning at B. Both {A,B,B',C} & {ABC, B'} are correct, there are no misassemblies or any other error in any of those representations.
This stage itself still makes sence in this case, since HairSplitter has it's own reassembly step, and it would be hard to connect B' with "part" of ABC without such splitting.
Also, some assemblers (not sure about Flye though) can output not only final contigs but unitigs or nodes of the underlying graph - is it what is really required from this step?
Because minigraph was used, possibly reads are aligned not to the assembly(contigs) but to the assembly _graph_? This would reduce my concerns about this step, although this step should be still evaluated separately, i.e. those breakpoints can be compared with quast-reported misassemblies.
Minor issues:
Line 23: metaMDBG is not HiFi only but work on the ONT too. It would be really benefitial to include this tool in all the comparisons.
Line 28: To the best of my knowledge, ONT has even bigger limitations regarding the quantity of DNA than hifi. Anyway, some reference is required here.
Line 83: closing bracket missing
Lines 107-112: It is not clear whether described clusterisation is limited by contigs' ends or not (I suppose it is), clarification needed.
Line 107: Minimap2 has different settings presets for hifi and ont (-x option) Were they actually meant as default? Or no options other than input/output and threads were used?
Line 103: What about structural variations? Major ones likely were splitted in assembly correction step, but how are minor one processed?
Line 136: is it the same k as above (5)? Anyway, further explanation of the chinese whispers algorithm would be helpful - i.e. can it only split some of the previous clusters or output something completely independent from that inital clusterisation?
Line 146: Racon ref is likely incorrect, I suppose it should be https://genome.cshlp.org/content/early/2017/01/18/gr.214270.116
Line 181: To show the tools performance on the real data it can be benefitial to use estimated strains coverage from some of the already studied datasets and not just 30,20,10,5. metaMDBG uses a zymo dataset with uneven coverage between species/strains (in the contrast to used in HairSplitter), it can be benefitial to compare on it
Supplementary result tables: would be also nice to have total assembly length there
Discussion: assembly/assembly graph alignment for assembly correction should clarified.




