
Latest recommendations
| Id | Title * | Authors * | Abstract * ▼ | Picture * | Thematic fields * | Recommender | Reviewers | Submission date | |
|---|---|---|---|---|---|---|---|---|---|
22 Jul 2024
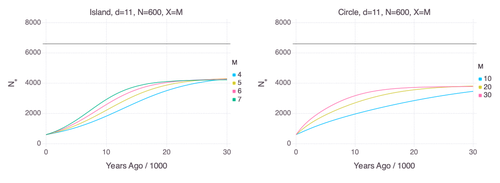
Genetic Evidence for Geographic Structure within the Neanderthal PopulationDecline in Neanderthal effective population size due to geographic structure and gene flowRecommended by Raquel Assis based on reviews by David Bryant and Guillaume AchazPublished PSMC estimates of Neanderthal effective population size (𝑁e) show an approximately five-fold decline over the past 20,000 years [1]. This observation may be attributed to a true decline in Neanderthal 𝑁e, statistical error that is notorious with PSMC estimation, or geographic subdivision and gene flow that has been hypothesized to occur within the Neanderthal population. Determining which of these factors contributes to the observed decline in Neanderthal 𝑁e is an important question that can provide insight into human evolutionary history. Though it is widely believed that the decline in Neanderthal 𝑁e is due to geographic subdivision and gene flow, no prior studies have theoretically examined whether these evolutionary processes can yield the observed pattern. In this paper [2], Rogers tackles this problem by employing two mathematical models to explore the roles of geographic subdivision and gene flow in the Neanderthal population. Results from both models show that geographic subdivision and gene flow can indeed result in a decline in 𝑁e that mirrors the observed decline estimated from empirical data. In contrast, Rogers argues that neither statistical error in PSMC estimates nor a true decline in 𝑁e are expected to produce the consistent decline in estimated 𝑁e observed across three distinct Neanderthal fossils. Statistical error would likely result in variation among these curves, whereas a true decline in 𝑁e would produce shifted curves due to the different ages of the three Neanderthal fossils. In summary, Rogers provides convincing evidence that the most reasonable explanation for the observed decline in Neanderthal 𝑁e is geographic subdivision and gene flow. Rogers also provides a basis for understanding this observation, suggesting that 𝑁e declines over time because coalescence times are shorter between more recent ancestors, as they are more likely to be geographic neighbors. Hence, Rogers’ theoretical findings shed light on an interesting aspect of human evolutionary history. References [1] Fabrizio Mafessoni, Steffi Grote, Cesare de Filippo, Svante Pääbo (2020) “A high-coverage Neandertal genome from Chagyrskaya Cave”. Proceedings of the National Academy of Sciences USA 117: 15132- 15136. https://doi.org/10.1073/pnas.2004944117 [2] Alan Rogers (2024) “Genetic evidence for geographic structure within the Neanderthal population”. bioRxiv, version 4 peer-reviewed and recommended by Peer Community in Mathematical and Computational Biology. https://doi.org/10.1101/2023.07.28.551046 | Genetic Evidence for Geographic Structure within the Neanderthal Population | Alan R. Rogers | <p>PSMC estimates of Neanderthal effective population size (N<sub>e</sub>)exhibit a roughly 5-fold decline across the most recent 20 ky before the death of each fossil. To explain this pattern, this article develops new theory relating... | | Evolutionary Biology, Genetics and population Genetics | Raquel Assis | 2023-10-17 18:06:38 | ||
08 Nov 2024
Bayesian joint-regression analysis of unbalanced series of on-farm trialsHandling Data Imbalance and G×E Interactions in On-Farm Trials Using Bayesian Hierarchical ModelsRecommended by Sophie Donnet based on reviews by Pierre Druilhet and David Makowski based on reviews by Pierre Druilhet and David Makowski
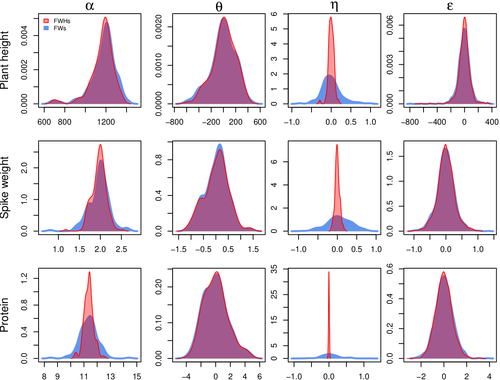
The article, "Bayesian Joint-Regression Analysis of Unbalanced Series of On-Farm Trials," presents a Bayesian statistical framework tailored for analyzing highly unbalanced datasets from participatory plant breeding (PPB) trials, specifically wheat trials. The key goal of this research is to address the challenges of genotype-environment (G×E) interactions in on-farm trials, which often have limited replication and varied testing conditions across farms. The study applies a hierarchical Bayesian model with Finlay-Wilkinson regression, which improves the estimation of G×E effects despite substantial data imbalance. By incorporating a Student’s t-distribution for residuals, the model is more robust to extreme values, which are common in on-farm trials due to variable environments. Note that the model allows a detailed breakdown of variance, identifying environment effects as the most significant contributors, thus highlighting areas for future breeding focus. Using Hamiltonian Monte Carlo methods, the study achieves reasonable computation times, even for large datasets. Obviously, the limitation of the methods comes from the level of data balance and replication. The method requires a minimum level of data balance and replication, which can be a challenge in very decentralized breeding networks Moreover, the Bayesian framework, though computationally feasible, may still be complex for widespread adoption without computational resources or statistical expertise. The paper presents a sophisticated Bayesian framework specifically designed to tackle the challenges of unbalanced data in participatory plant breeding (PPB). It showcases a novel way to manage the variability in on-farm trials, a common issue in decentralized breeding programs. This study's methods accommodate the inconsistencies inherent in on-farm trials, such as extreme values and minimal replication. By using a hierarchical Bayesian approach with a Student’s t-distribution for robustness, it provides a model that maintains precision despite these real-world challenges. This makes it especially relevant for those working in unpredictable agricultural settings or decentralized trials. From a more general perspective, this paper’s findings support breeding methods that prioritize specific adaptation to local conditions. It is particularly useful for researchers and practitioners interested in breeding for agroecological or organic farming systems, where G×E interactions are critical but hard to capture in traditional trial setups. Beyond agriculture, the paper serves as an excellent example of advanced statistical modeling in highly variable datasets. Its applications extend to any field where data is incomplete or irregular, offering a clear case for hierarchical Bayesian methods to achieve reliable results. Finally, although begin quite methodological, the paper provides practical insights into how breeders and researchers can work with farmers to achieve meaningful varietal evaluations. References Michel Turbet Delof , Pierre Rivière , Julie C Dawson, Arnaud Gauffreteau , Isabelle Goldringer , Gaëlle van Frank , Olivier David (2024) Bayesian joint-regression analysis of unbalanced series of on-farm trials. HAL, ver.2 peer-reviewed and recommended by PCI Math Comp Biol https://hal.science/hal-04380787 | Bayesian joint-regression analysis of unbalanced series of on-farm trials | Michel Turbet Delof , Pierre Rivière , Julie C Dawson, Arnaud Gauffreteau , Isabelle Goldringer , Gaëlle van Frank , Olivier David | <p>Participatory plant breeding (PPB) is aimed at developing varieties adapted to agroecologically-based systems. In PPB, selection is decentralized in the target environments, and relies on collaboration between farmers, farmers' organisations an... | | Agricultural Science, Genetics and population Genetics, Probability and statistics | Sophie Donnet | Pierre Druilhet, David Makowski | 2024-01-11 14:17:41 | |
24 Dec 2020
A linear time solution to the Labeled Robinson-Foulds Distance problemComparing reconciled gene trees in linear timeRecommended by Céline Scornavacca based on reviews by Barbara Holland, Gabriel Cardona, Jean-Baka Domelevo Entfellner and 1 anonymous reviewer
Unlike a species tree, a gene tree results not only from speciation events, but also from events acting at the gene level, such as duplications and losses of gene copies, and gene transfer events [1]. The reconciliation of phylogenetic trees consists in embedding a given gene tree into a known species tree and, doing so, determining the location of these gene-level events on the gene tree [2]. Reconciled gene trees can be seen as phylogenetic trees where internal node labels are used to discriminate between different gene-level events. Comparing them is of foremost importance in order to assess the performance of various reconciliation methods (e.g. [3]). References [1] Maddison, W. P. (1997). Gene trees in species trees. Systematic biology, 46(3), 523-536. doi: https://doi.org/10.1093/sysbio/46.3.523 | A linear time solution to the Labeled Robinson-Foulds Distance problem | Samuel Briand, Christophe Dessimoz, Nadia El-Mabrouk and Yannis Nevers | <p>Motivation Comparing trees is a basic task for many purposes, and especially in phylogeny where different tree reconstruction tools may lead to different trees, likely representing contradictory evolutionary information. While a large variety o... | | Combinatorics, Design and analysis of algorithms, Evolutionary Biology | Céline Scornavacca | 2020-08-20 21:06:23 | ||
27 Jan 2025
Biology-Informed inverse problems for insect pests detection using pheromone sensorsTowards accurate inference of insect presence landscapes from pheromone sensor networksRecommended by Eric Tannier based on reviews by Angelo Iollo and 1 anonymous reviewer
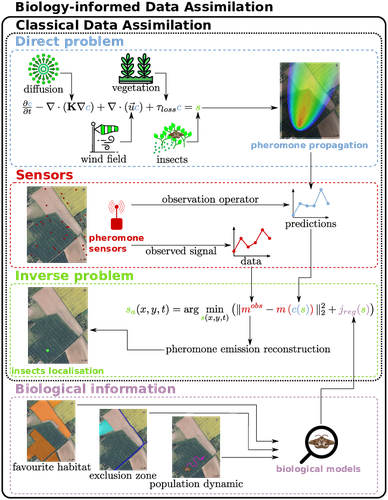
Insecticides are used to control crop pests and prevent severe crop losses. They are also a major cause of the current decline in biodiversity, contribute to climate change, and pollute soil and water, with consequences for human and environmental health [1]. The rationale behind the work of Malou et al [2] is that some pesticide application protocols can be improved by a better knowledge of the insects, their biology, their ecology and their real-time infestation dynamics in the fields. Thanks to a network of pheromone sensors and a mathematical method to derive the spatio-temporal distribution of pest populations from the signals, it is theoretically possible to adjust the time, dose and area of treatment and to use less pesticide with greater efficiency than an uninformed protocol. Malou et al [2] focus on the mathematical problem, recognising that its real role in pest control would require work on its implementation and on a benefit-harm analysis. The problem is an "inverse problem" [3] in that it consists of inferring the presence of insects from the trail left by the pheromones, given a model of pheromone diffusion by insects. The main contribution of this work is the formulation and comparison of different regularisation terms in the optimisation inference scheme, in order to guide the optimisation by biological knowledge of specific pests, such as some parameters of population dynamics. The accuracy and precision of the results are tested and compared on a simple toy example to test the ability of the model and algorithm to detect the source of the pheromones and the efficiency of the data assimilation principle. A further simulation is then carried out on a real plot with realistic parameters and rules based on knowledge of a maize pest. A repositioning of the sensors (informed by the results from the initial positions) is carried out during the test phase to allow better detection. The work of Malou et al [2] is large, deep and complete. Its includes a detailed study of the numerical solutions of different data assimilation methods, as well as a theoretical reflection on how this work could contribute to agricultural and environmental issues. References [1] IPBES (2024). Thematic Assessment Report on the Underlying Causes of Biodiversity Loss and the Determinants of Transformative Change and Options for Achieving the 2050 Vision for Biodiversity of the Intergovernmental Science-Policy Platform on Biodiversity and Ecosystem Services. O’Brien, K., Garibaldi, L., and Agrawal, A. (eds.). IPBES secretariat, Bonn, Germany. https://doi.org/10.5281/zenodo.11382215 [2] Thibault Malou, Nicolas Parisey, Katarzyna Adamczyk-Chauvat, Elisabeta Vergu, Béatrice Laroche, Paul-Andre Calatayud, Philippe Lucas, Simon Labarthe (2025) Biology-Informed inverse problems for insect pests detection using pheromone sensors. HAL, ver.2 peer-reviewed and recommended by PCI Math Comp Biol https://hal.inrae.fr/hal-04572831v2 [3] Isakov V (2017). Inverse Problems for Partial Differential Equations. Vol. 127. Applied Mathematical Sciences. Cham: Springer International Publishing. https://doi.org/10.1007/978-3-319-51658-5. | Biology-Informed inverse problems for insect pests detection using pheromone sensors | Thibault Malou, Nicolas Parisey, Katarzyna Adamczyk-Chauvat, Elisabeta Vergu, Béatrice Laroche, Paul-Andre Calatayud, Philippe Lucas, Simon Labarthe | <p>Most insects have the ability to modify the odor landscape in order to communicate with their conspecies during key phases of their life cycle such as reproduction. They release pheromones in their nearby environment, volatile compounds that ar... | | Agricultural Science, Dynamical systems, Epidemiology, Systems biology | Eric Tannier | 2024-05-12 19:14:34 | ||
02 Oct 2024
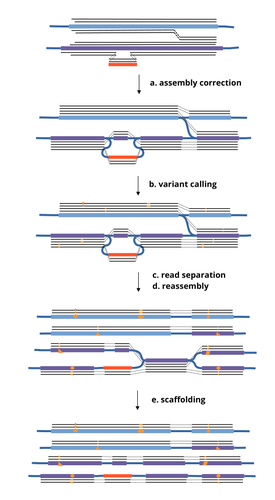
HairSplitter: haplotype assembly from long, noisy readsAccurate Haplotype Reconstruction from Long, Error-Prone, Reads with HairSplitterRecommended by Giulio Ermanno Pibiri based on reviews by Dmitry Antipov and 1 anonymous reviewer
A prominent challenge in computational biology is to distinguish microbial haplotypes -- closely related organisms with highly similar genomes -- due to small genomic differences that can cause significant phenotypic variations. Current genome assembly tools struggle with distinguishing these haplotypes, especially for long-read sequencing data with high error rates, such as PacBio or Oxford Nanopore Technology (ONT) reads. While existing methods work well for either viral or bacterial haplotypes, they often fail with low-abundance haplotypes and are computationally intensive. This work by Faure, Lavenier, and Flot [1] introduces a new tool -- HairSplitter -- that offers a solution for both viral and bacterial haplotype separation, even with error-prone long reads. It does this by efficiently calling variants, clustering reads into haplotypes, creating new separated contigs, and resolving the assembly graph. A key advantage of HairSplitter is that it is entirely parameter-free and does not require prior knowledge of the organism's ploidy. HairSplitter is designed to handle both metaviromes and bacterial metagenomes, offering a more versatile and efficient solution than existing tools, like stRainy [2], Strainberry [3], and hifiasm-meta [4]. References [1] Roland Faure, Dominique Lavenier, Jean-François Flot (2024) HairSplitter: haplotype assembly from long, noisy reads. bioRxiv, ver.3 peer-reviewed and recommended by PCI Math Comp Biol https://doi.org/10.1101/2024.02.13.580067 [2] Kazantseva E, A Donmez, M Pop, and M Kolmogorov (2023). stRainy: assembly-based metagenomic strain phasing using long reads. Bioinformatics. https://doi.org/10.1101/2023.01.31.526521 [3] Vicedomini R, C Quince, AE Darling, and R Chikhi (2021). Strainberry: automated strain separation in low complexity metagenomes using long reads. Nature Communications, 12, 4485. ISSN: 2041-1723. https://doi.org/10.1038/s41467-021-24515-9 [4] Feng X, H Cheng, D Portik, and H Li (2022). Metagenome assembly of high-fidelity long reads with hifiasm-meta. Nature Methods, 19, 1–4. https://doi.org/10.1038/s41592-022-01478-3 | HairSplitter: haplotype assembly from long, noisy reads | Roland Faure, Dominique Lavenier, Jean-François Flot | <p>Long-read assemblers face challenges in discerning closely related viral or<br>bacterial strains, often collapsing similar strains in a single sequence. This limitation has<br>been hampering metagenome analysis, where diverse strains may harbor... | | Design and analysis of algorithms, Development, Genomics and Transcriptomics, Probability and statistics | Giulio Ermanno Pibiri | 2024-02-15 10:17:04 | ||
30 Mar 2025
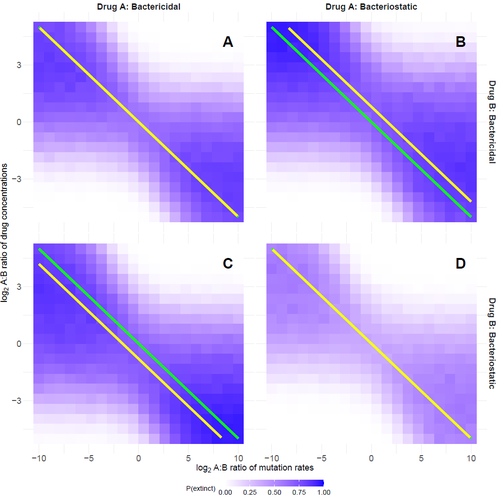
Optimal antimicrobial dosing combinations when drug-resistance mutation rates differOptimizing antibiotic use: How much should we favor drugs with lower resistance mutation rates?Recommended by Amaury Lambert based on reviews by 2 anonymous reviewersIn the hereby recommended paper [1], Delaney, Letten and Engelstädter study the appearance of antibiotic resistance(s) in a bacterial population subject to a combination of two antibiotics \( A \) and \( B \), in concentrations \( C_A \) and \( C_B \) respectively. Their goal was to find optimal values of \( C_A \) and \( C_B \) that minimize the risk of evolutionary rescue, under the unusual assumption that resistance mutations to either antibiotic are not equally likely. The authors introduce a stochastic model assuming that the susceptible population grows like a supercritical birth-death process which becomes subcritical in the presence of antibiotics (and exactly critical for a certain concentration c of a single antibiotic): the effect of each antibiotic is either to reduce division rate (bacteriostatic drug) or to enhance death rate (bacteriocidal drug) by a factor which has a sigmoid functional dependence on antibiotic concentration. Now at each division, a resistance mutation can arise, with probability \( \mu_A \) for a resistance to antibiotic \( A \) and with probability \( \mu_B \) for a resistance to antibiotic \( B \). The goal of the paper is to find the optimal ratio of drug concentrations \( C_A \) and \( C_B \) when these are subject to a constrain \( C_A + C_B = c \), depending on the ratio of mutation rates. Assuming total resistance and no cross resistance, the authors show that the optimal concentrations are given by \( C_A = c/(1+\sqrt{\mu_A/\mu_B }) \) and \( C_B = c/(1+\sqrt{\mu_B/\mu_A}) \), which leads to the beautiful result that the optimal ratio \( C_A/C_B \) is equal to the square root of \( \mu_B/\mu_A \). The authors have made a great job completing their initial submission by simulations of model extensions, relaxing assumptions like single antibiotic mode, absence of competition, absence of cost of resistance, sharp cutoff in toxicity… and comparing the results obtained by simulation to their mathematical result. The paper is very clearly written and any reader interested in antibiotic resistance, stochastic modeling of bacterial populations and/or evolutionary rescue will enjoy reading it. Let me thank the authors for their patience and for their constant willingness to comply with the reviewers’ and recommender’s demands during the reviewing process.
Reference Oscar Delaney, Andrew D. Letten, Jan Engelstaedter (2025) Optimal antimicrobial dosing combinations when drug-resistance mutation rates differ. bioRxiv, ver.3 peer-reviewed and recommended by PCI Mathematical and Computational Biology https://doi.org/10.1101/2024.05.04.592498 | Optimal antimicrobial dosing combinations when drug-resistance mutation rates differ | Oscar Delaney, Andrew D. Letten, Jan Engelstaedter | <p>Given the ongoing antimicrobial resistance crisis, it is imperative to develop dosing regimens optimised to avoid the evolution of resistance. The rate at which bacteria acquire resistance-conferring mutations to different antimicrobial drugs s... | | Evolutionary Biology | Amaury Lambert | 2024-05-07 17:17:55 | ||
10 Jan 2024
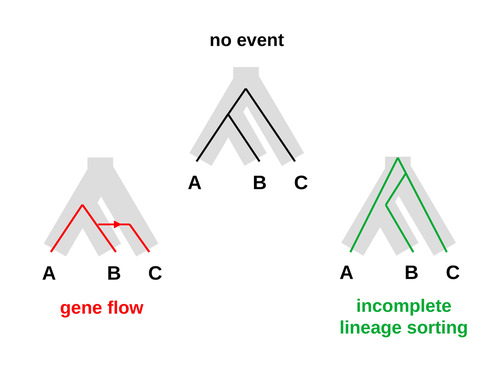
An approximate likelihood method reveals ancient gene flow between human, chimpanzee and gorillaAphid: A Novel Statistical Method for Dissecting Gene Flow and Lineage Sorting in Phylogenetic ConflictRecommended by Alan Rogers based on reviews by Richard Durbin and 2 anonymous reviewers
Galtier [1] introduces “Aphid,” a new statistical method that estimates the contributions of gene flow (GF) and incomplete lineage sorting (ILS) to phylogenetic conflict. Aphid is based on the observation that GF tends to make gene genealogies shorter, whereas ILS makes them longer. Rather than fitting the full likelihood, it models the distribution of gene genealogies as a mixture of several canonical gene genealogies in which coalescence times are set equal to their expectations under different models. This simplification makes Aphid far faster than competing methods. In addition, it deals gracefully with bidirectional gene flow—an impossibility under competing models. Because of these advantages, Aphid represents an important addition to the toolkit of evolutionary genetics. In the interest of speed, Aphid makes several simplifying assumptions. Yet even when these were violated, Aphid did well at estimating parameters from simulated data. It seems to be reasonably robust. Aphid studies phylogenetic conflict, which occurs when some loci imply one phylogenetic tree and other loci imply another. This happens when the interval between successive speciation events is fairly short. If this interval is too short, however, Aphid’s approximations break down, and its estimates are biased. Galtier suggests caution when the fraction of discordant phylogenetic trees exceeds 50–55%. Thus, Aphids will be most useful when the interval between speciation events is short, but not too short. Galtier applies the new method to three sets of primate data. In two of these data sets (baboons and African apes), Aphid detects gene flow that would likely be missed by competing methods. These competing methods are primarily sensitive to gene flow that is asymmetric in two senses: (1) greater flow in one direction than the other, and (2) unequal gene flow connecting an outgroup to two sister species. Aphid finds evidence of symmetric gene flow in the ancestry of baboons and also in that of African apes. The data suggest that ancestral humans and chimpanzees both interbred with ancestral gorillas, and at about the same rate. Aphid’s ability to detect this signature sets it apart from competing methods. References [1] Nicolas Galtier (2023) “An approximate likelihood method reveals ancient gene flow between human, chimpanzee and gorilla”. bioRxiv, ver. 3 peer-reviewed and recommended by Peer Community in Mathematical and Computational Biology. https://doi.org/10.1101/2023.07.06.547897 | An approximate likelihood method reveals ancient gene flow between human, chimpanzee and gorilla | Nicolas Galtier | <p>Gene flow and incomplete lineage sorting are two distinct sources of phylogenetic conflict, i.e., gene trees that differ in topology from each other and from the species tree. Distinguishing between the two processes is a key objective of curre... | | Evolutionary Biology, Genetics and population Genetics, Genomics and Transcriptomics | Alan Rogers | 2023-07-06 18:41:16 | ||
26 Feb 2024

A workflow for processing global datasets: application to intercroppingCollecting, assembling and sharing data in crop sciencesRecommended by Eric Tannier based on reviews by Christine Dillmann and 2 anonymous reviewers
It is often the case that scientific knowledge exists but is scattered across numerous experimental studies. Because of this dispersion in different formats, it remains difficult to access, extract, reproduce, confirm or generalise. This is the case in crop science, where Mahmoud et al [1] propose to collect and assemble data from numerous field experiments on intercropping. It happens that the construction of the global dataset requires a lot of time, attention and a well thought-out method, inspired by the literature on data science [2] and adapted to the specificities of crop science. This activity also leads to new possibilities that were not available in individual datasets, such as the detection of full factorial designs using graph theory tools developed on top of the global dataset. The study by Mahmoud et al [1] has thus multiple dimensions:
I was particularly interested in the promotion of the FAIR principles, perhaps used a little too uncritically in my view, as an obvious solution to data sharing. On the one hand, I am admiring and grateful for the availability of these data, some of which have never been published, nor associated with published results. This approach is likely to unearth buried treasures. On the other hand, I can understand the reluctance of some data producers to commit to total, definitive sharing, facilitating automatic reading, without having thought about a certain reciprocity on the part of users and use by artificial intelligence. Reciprocity in terms of recognition, as is discussed by Mahmoud et al [1], but also in terms of contribution to the commons [5] or reading conditions for machine learning. References [1] Mahmoud R., Casadebaig P., Hilgert N., Gaudio N. A workflow for processing global datasets: application to intercropping. 2024. ⟨hal-04145269v2⟩ ver. 2 peer-reviewed and recommended by Peer Community in Mathematical and Computational Biology. https://hal.science/hal-04145269 [2] Wickham, H. 2014. Tidy data. Journal of Statistical Software 59(10) https://doi.org/10.18637/jss.v059.i10 [3] Gaudio, N., R. Mahmoud, L. Bedoussac, E. Justes, E.-P. Journet, et al. 2023. A global dataset gathering 37 field experiments involving cereal-legume intercrops and their corresponding sole crops. https://doi.org/10.5281/zenodo.8081577 [4] Mahmoud, R., Casadebaig, P., Hilgert, N. et al. Species choice and N fertilization influence yield gains through complementarity and selection effects in cereal-legume intercrops. Agron. Sustain. Dev. 42, 12 (2022). https://doi.org/10.1007/s13593-022-00754-y [5] Bernault, C. « Licences réciproques » et droit d'auteur : l'économie collaborative au service des biens communs ?. Mélanges en l'honneur de François Collart Dutilleul, Dalloz, pp.91-102, 2017, 978-2-247-17057-9. https://shs.hal.science/halshs-01562241 | A workflow for processing global datasets: application to intercropping | Rémi Mahmoud, Pierre Casadebaig, Nadine Hilgert, Noémie Gaudio | <p>Field experiments are a key source of data and knowledge in agricultural research. An emerging practice is to compile the measurements and results of these experiments (rather than the results of publications, as in meta-analysis) into global d... | | Agricultural Science | Eric Tannier | 2023-06-29 15:38:28 | ||
28 Jun 2024
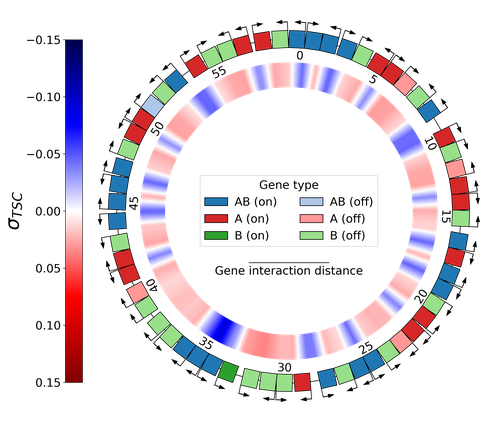
Emergence of Supercoiling-Mediated Regulatory Networks through the Evolution of Bacterial Chromosome OrganizationUnderstanding the impact of the transcription-supercoiling coupling on bacterial genome evolutionRecommended by Nelle Varoquaux based on reviews by Ivan Junier and 1 anonymous reviewer
DNA supercoiling, the under or overwinding of DNA, is known to strongly impact gene expression, as changes in levels of supercoiling directly influence transcription rates. In turn, gene transcription generates DNA supercoiling on each side of an advancing RNA polymerase. This coupling between DNA supercoiling and transcription may result in different outcomes, depending on neighboring gene orientations: divergent genes tend to increase transcription levels, convergent genes tend to inhibit each other, while tandem genes may exhibit more intricate relationships. While several works have investigated the relationship between transcription and supercoiling, Grohens et al [1] address a different question: how does transcription-supercoiling coupling drive genome evolution? To this end, they consider a simple model of gene expression regulation where transcription level only depends on the local DNA supercoiling and where the transcription of one gene generates a linear profile of positive and negative DNA supercoiling on each side of it. They then make genomes evolve through genomic inversions only considering a fitness that reflects the ability of a genome to cope with two distinct environments for which different genes have to be activated or repressed. Using this simple model, the authors illustrate how evolutionary adaptation via genomic inversions can adjust expression levels for enhanced fitness within specific environments, particularly with the emergence of relaxation-activated genes. Investigating the genomic organization of individual genomes revealed that genes are locally organized to leverage the transcription-supercoiling coupling for activation or inhibition, but larger-scale networks of genes are required to strongly inhibit genes (sometimes up to networks of 20 genes). Thus, supercoiling-mediated interactions between genes can implicate more than just local genes. Finally, they construct an "effective interaction graph" between genes by successively simulating gene knock-outs for all of the genes of an individual and observing the effect on the expression level of other genes. They observe a densely connected interaction network, implying that supercoiling-based regulation could evolve concurrently with genome organization in bacterial genomes. References [1] Théotime Grohens, Sam Meyer, Guillaume Beslon (2024) Emergence of Supercoiling-Mediated Regulatory Networks through the Evolution of Bacterial Chromosome Organization. bioRxiv, ver. 4 peer-reviewed and recommended by Peer Community in Mathematical and Computational Biology https://doi.org/10.1101/2022.09.23.509185 | Emergence of Supercoiling-Mediated Regulatory Networks through the Evolution of Bacterial Chromosome Organization | Théotime Grohens, Sam Meyer, Guillaume Beslon | <p>DNA supercoiling -- the level of twisting and writhing of the DNA molecule around itself -- plays a major role in the regulation of gene expression in bacteria by modulating promoter activity. The level of DNA supercoiling is a dynamic property... | | Biophysics, Evolutionary Biology, Systems biology | Nelle Varoquaux | 2023-06-30 10:34:28 | ||
27 Sep 2024
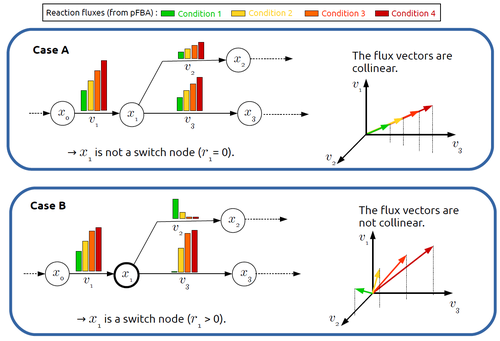
In silico identification of switching nodes in metabolic networksA computational method to identify key players in metabolic rewiringRecommended by Claudine Chaouiya based on reviews by 2 anonymous reviewers
Significant progress has been made in developing computational methods to tackle the analysis of the numerous (genome-wide scale) metabolic networks that have been documented for a wide range of species. Understanding the behaviours of these complex reaction networks is crucial in various domains such as biotechnology and medicine. Metabolic rewiring is essential as it enables cells to adapt their metabolism to changing environmental conditions. Identifying the metabolites around which metabolic rewiring occurs is certainly useful in the case of metabolic engineering, which relies on metabolic rewiring to transform micro-organisms into cellular factories [1], as well as in other contexts. This paper by F. Mairet [2] introduces a method to disclose these metabolites, named switch nodes, relying on the analysis of the flux distributions for different input conditions. Basically, considering fluxes for different inputs, which can be computed using e.g. Parsimonious Flux Balance Analysis (pFBA), the proposed method consists in identifying metabolites involved in reactions whose different flux vectors are not collinear. The approach is supported by four case studies, considering core and genome-scale metabolic networks of Escherichia coli, Saccharomyces cerevisiae and the diatom Phaeodactylum tricornutum. Whilst identified switch nodes may be biased because computed flux vectors satisfying given objectives are not necessarily unique, the proposed method has still a relevant predictive potential, complementing the current array of computational methods to study metabolism. References [1] Tao Yu, Yasaman Dabirian, Quanli Liu, Verena Siewers, Jens Nielsen (2019) Strategies and challenges for metabolic rewiring. Current Opinion in Systems Biology, Vol 15, pp 30-38. https://doi.org/10.1016/j.coisb.2019.03.004. [2] Francis Mairet (2024) In silico identification of switching nodes in metabolic networks. bioRxiv, ver.3 peer-reviewed and recommended by PCI Math Comp Biol https://doi.org/10.1101/2023.05.17.541195 | In silico identification of switching nodes in metabolic networks | Francis Mairet | <p>Cells modulate their metabolism according to environmental conditions. A major challenge to better understand metabolic regulation is to identify, from the hundreds or thousands of molecules, the key metabolites where the re-orientation of flux... | | Graph theory, Physiology, Systems biology | Claudine Chaouiya | Anonymous | 2023-05-26 17:24:26 |




